TLDR: Nicotine is mostly wrongly vilified; its big issue is a short half-life that foments chronic re-dosing. Addiction potential is a function of net time to hit your bloodstream (inhalation/smoking > buccal administration/snus/dip > nicotine gums > transdermal patches). Be careful with chronically elevated nicotine levels, which can engender higher baseline levels of neural excitation (mechanisms explicated below) and is a primary mediator of addiction reinforcement. Otherwise, it’s a powerful, angiogenic nootropic that is useful with perspicacious use. But as always, you’re modifying a vastly complex biochemical system with countless reflexive feedback loops, so who knows.
If you refer to my admittedly indulgent list of “apodictic truths”, I make the point that “(d)opaminergics are a dual-edged sword and extreme caution is warranted.” While nicotine is more rightly classified as a cholinergic, it does elicit a strong dopamine response via mechanisms downstream of nicotinic acetylcholine receptor (nAChR) activation. This ultimately begets dopamine tolerance and an attendant progressive attenuation of reward-prediction error in the mesolimbic pathway. If you want the mechanistic details, read the wikipedia page or, better yet, the Examine.com page.
A few relevant details on nicotine pharmacokinetics:
- Half-life: 1-2 hours
- Duration of Action: .5-1.5 hour (typically contingent on tolerance in naive vs chronic users)
- Metabolism: mostly in the liver via CYP2A6, CYP2B6, FMO3
- Major metabolite: Cotinine (also a nAChR agonist w/ a weaker affinity and ~20 hour half-life)
Source link
Nicotine is now back in vogue owing to widespread vape use in the exoterica. In the esoterica, nicotine is consistently rated relatively highly on nootropics user surveys. It is mostly utilized as a short-acting stimulant.
Gwern has a fairly exhaustive lit-review-cum-exegesis on nicotine that’s worth a read; the upshot is that infrequent and/or intermittent nicotine gum usage is useful, relatively safe, and not detrimentally habit-forming. He concludes that vaping should be avoided too. That’s also my conclusion on an independent reading of the relevant literature and with personal experimentation.
Popular podcasts like Peter Attia’s Drive and the Huberman Lab have also given treatment to the topic. I personally weigh the former much more heavily— the Huberman Lab podcast had about 10 great episodes in the eponymous host’s wheelhouse before devolving into a vortex of incessant conjecture and caveating. I’m at the Gell-Mann hypermnesia stage after he’s given perfunctory and error-prone reports on areas I know well. (My recommendations: don’t take magnesium glycine, mag l-threonate, or apigenin for sleep). Tangentially, Emil Kirkegaard also has an interesting analysis on whether vaping will shrink your balls. Spoiler alert: probably not.
On to the biochemistry: nAChR’s are inotropic cation (sodium, potassium, calcium) channels with various subunit configurations that are somewhat similar to GABA(A) receptor subunits, which form inotropic anion (mostly chloride and some bicarbonate ions) channels. When activated by their respective ligands, the former adjusts neuronal surface membrane potential such that the cells are more likely to depolarize when cell signals propagate, the latter does the opposite: creating a diathesis towards hyperpolarization. Both receptor types can further influence intracellular cascades and gene transcription. Broadly, nAChR activation increases attention, focus, and drive, though transiently.
nAChR’s are located through the body and are involved in autonomic and somatic functions. As an aside, many nerve agents like Novichok are acetylcholinesterase (the enzyme that breaks down acetylcholine) inhibitors at the neuromuscular junction. This eventually brings about a fulminant cholinergic crisis whereby autonomic respiratory and cardiac functions are arrested, and a victim will die.
Anyways, we’ll focus on two receptor subtypes located in the CNS/brain that are germane to nicotine activation:
- (α4)2(β2)3
- (α7)5
The first is expressed at a higher density in GABAergic neurons (activation of these neurons elicits GABA release synaptically and—as an epiphenomenon—extra-synaptically as well to mediate tonic inhibition). α4-containing AChRs are desensitized to ligands like nicotine rapidly (via receptor internalization typically) and only re-sensitize after ~1 hour, though this is obviously person-dependent. Conversely, α7-containing AChRs desensitize much more slowly and re-sensitize on the order of ~2 minutes. A principal difference is that α7-containing AChRs are present in higher densities on glutaminergic neurons extending from the ventral tegmental area (VTA) to the prefrontal cortex (PFC), and α4-containing AChRs are primarily expressed on afferent and interneuron GABAergic cells extending from the nucleus accumbens (NAcc) to the VTA. See the diagram below taken from this 2002 paper in Neuron.
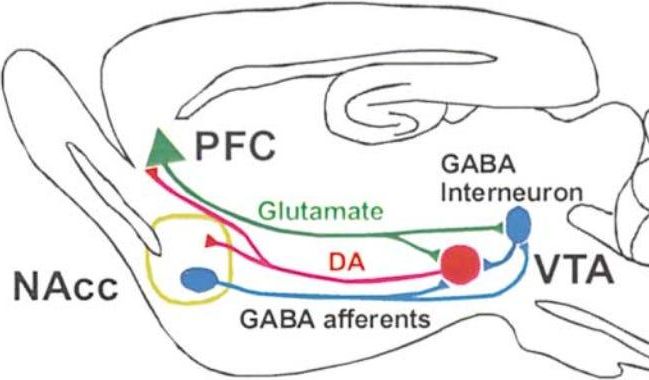
So what’s the problem? The problem is that with recurrent nicotine dosing, α4-containing AChRs are activated and rapidly desensitized, leading to a transient spike in GABA release (net inhibitory, anxiolytic). Activation of α7-containing AChR, on the obverse, causes a more durable spike in glutamate (net excitatory, anxiogenic) (see the upper right hand graph and diagram below). The ‘LTP’ in the figure stands for “long term potentiation”. With chronically elevated levels as with smoking/vaping/dipping (or especially ebullient Nicorette chewing), the baseline firing threshold is increased (think about the membrane potential with a higher propensity to depolarize and fire that we spoke about earlier).
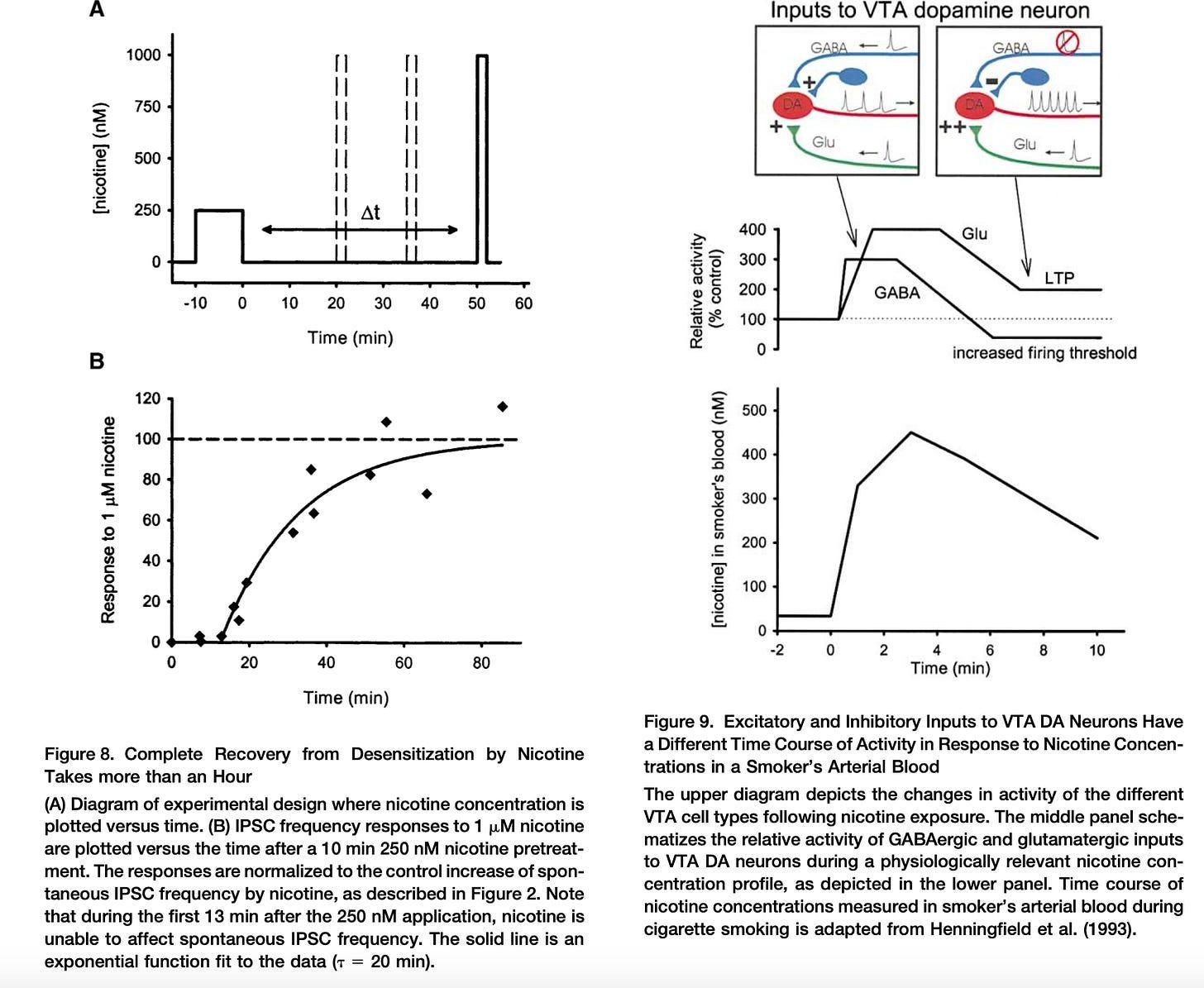
Source here
So now we have a picture where the chronic user is re-dosing chronically for both the mental stimulation and the anxiolytic component of ephemerally elevated GABA release. This is the so-called Nesbitt’s Paradox — that cigarette smoking generates physiological and psychological changes which are normally incompatible, namely increased arousal together with decreased stress. The user-level reality is much more grim: chronic users are grasping at a baseline that has long been altered. In some ways this is an affine relationship to alcohol tolerance — GABA(a) receptors are down-regulated in response to constitutive agonist binding and baseline neuronal excitability increases as a compensatory response. Curiously, alcohol has also been shown to be a nAChR positive allosteric modulator, which is a bit counterintuitive for the canonical depressant. But alcohol has pleiotropic, promiscuous, and systemic intra- and extracellular effects that have yet to be fully characterized.
This goes beyond the scope of this post, but many of the compensatory baseline changes in both alcohol and nicotine addiction models are likely mediated by ΔFosB-related epigenetic changes that down-regulate the transcription of receptor proteins like α4-containing AChRs (or α4-containing GABA(a) receptors in the case of alcohol tolerance). What’s particularly pernicious about this transcription factor is that its induction can create a positive feedback loop. Per wikipedia, “Repression of c-Fos by ΔFosB, which consequently further induces expression of ΔFosB, forms a positive feedback loop that serves to indefinitely perpetuate the addictive state.”
So, the primary takeaway is if you’re going to use nicotine, use it on a pro re nata and intermittent basis (and not ‘intermittent’ in the current demotic sense, but rather spasmodically). Some people will go as far as to say that caffeine is more addictive than nicotine so regular use is OK. This isn’t true as measured by dopamine-evoked currents, but when dose-adjusted it isn’t manifestly untrue. Here idiosyncratic personal response is relevant, but it’s also important to note that nicotine’s comparatively short duration of action makes it more amenable to abuse. An interesting corollary is that in “fast” caffeine metabolizers, the CYP1A2 enzyme (metabolizes caffeine) is highly inducible by nicotine. Per SNPedia, “In smokers, … the A/A homozygotes had 1.6x higher CYP1A2 activity than A/C and C/C genotypes”. This could conceivably shorten caffeine’s half-life to 1.5-2 hours with current nicotine use. I bear the highly inducible rs762551(A) allele, and it does subjectively seem like nicotine catalyzes faster caffeine clearance.
Secondary Considerations
I have a few other ancillary concerns about nicotine that I’ve enumerated below:
- LTP with chronic use could lead to glutamate-induced excitotoxicity and could very plausibly affect sleep architecture.
- Nicotine induces a transient prolactin release, which returns to baseline after ~2 hours. This is odd as dopamine inhibits prolactin release from the hypothalamus and the two tend to be inversely correlated.
- Testosterone may be affected by nicotine. Rat studies show a drop in serum testosterone after chronic use, but correlational human studies show that smokers tend to have higher levels of circulating testosterone. See below for speculation on why.
- Complex interactions with estrogen and aromatase: per Examine:
The ability of Nicotine (and related nicotine alkaloids, when looking at smoking studies) to inhibit the Aromatase enzyme may cause a shift towards androgens rather than estrogens over time. The degree of change noted in these studies may be higher than can be accredited to nicotine due to other tobacco alkaloids … Nicotine appears to act as an estrogen receptor antagonist, and can inhibit the signaling pathways of estrogen; It shows preference for ER-ß rather than the alpha subunit (the latter being associated with classical 'effects of estrogen'). The estrogen receptor subunit inhibited is the one associated with positive effects in both genders - Cotinine (~20 hour half-life) and nicotine probably inhibit 3α-hydroxysteroid dehydrogenase (3α-HSD) fairly potently. This is an crucial enzyme in the metabolism of dihydrotestosterone (DHT, or something that you’re likely concerned about vis-a-vis hair loss), but more importantly it also synthesizes critical neurosteroids like allopregnanolone (a GABA(A) positive allosteric modulator and a nAChR negative allosteric modulator).